



 PEXELS/babydov
PEXELS/babydov Few technologies have had a more profound impact on society than pharmaceuticals (drugs for medical use). The arrival of antibiotics has halted massive numbers of deaths from infection. Vaccines have made what were once leading causes of death now easily preventable. HIV antiretrovirals have turned a fatal condition into a manageable one. Oral contraceptives have provided women with tools to manage their fertility, with downstream effects on their lives and livelihoods. Emerging technologies, including gene therapies and personalized medicine, will continue to transform public health and society. Yet, not all societal impacts are intended: for example, the fetal harms caused by thalidomide, the rise of antibiotic resistance, and the role of pharmaceutical opioids in exacerbating this century’s opioid overdose crisis.
Few technologies are more ubiquitous than pharmaceuticals. There are more than 24,000 prescription drugs approved for marketing in the United States [1] and over 100,000 marketed nonprescription (over-the-counter) drugs [2]. About 50% of people in the United States use at least one prescription drug in a given month, with about 13% using five or more [3]. For people aged 65 and older, these rates are 90% and 43%, respectively [4]. In 2024, the United States spent over $800 B on prescription drugs in retail, clinic, and hospital pharmacy settings [5] and $44 B in over-the-counter retail sales [6].
Furthermore, few technologies are as tightly regulated and thoroughly scientifically evaluated as pharmaceuticals. In the United States, this mandate falls within the purview of the Food and Drug Administration (FDA). Examples of jurisdictions globally include the European Medicines Agency (EMA), Health Canada, and Japan’s Pharmaceuticals and Medical Devices Agency (PMDA). Like its counterparts, FDA promulgates detailed regulations about generating scientific evidence, securing marketing approval, and monitoring marketed pharmaceuticals.1
Pharmaceutical sciences—toxicology, pharmacology, clinical, biostatistics, chemistry, behavioral, and more—are the foundation of drug development and evaluation. But what brings these sciences together to inform regulatory judgments? For this, regulators have adapted universal risk assessment and decision science approaches to fit their specific decision-making needs. This includes benefit–Risk assessment to support a determination of whether the favorable effects (i.e., benefits) of a drug, in a defined context of use, exceed the unfavorable effects (i.e., risks) of that drug, accounting for important uncertainties.
This commentary offers societal insight about pharmaceutical benefit–Risk assessment to a broader technology audience. One aim is to demonstrate that pharmaceutical benefit–Risk assessment aligns with principles of risk governance for technologies with consequential public impact. Another is to elevate appreciation of the complex science and the nuanced judgments involved in these assessments. A third is to make core concepts of pharmaceutical benefit–Risk more accessible, recognizing their personal relevance to all of us who use medicine. My commentary draws very closely on FDA’s 2023 Guidance to Industry: Benefit–Risk Assessment for New Drugs and Biological Products [7] and worked examples, and it uses FDA’s phrasing where useful to ensure preserving FDA’s meaning. However, any interpretation reflects my personal perspective solely.
Regulatory Context for Benefit–Risk Assessment
The 1938 Food, Drug, and Cosmetic (FD&C) Act charges FDA with protecting public health by ensuring that drugs are safe and effective for their intended use, are of sufficient quality, are adequately labeled, and that information and marketing about drugs are not false or misleading. Entire articles could be written about each one of these pillars. This article focuses on the determination of “safe and effective,” and even more narrowly within the context of branded prescription drugs that are “new”2or an existing branded prescription drug that is proposed for a “new” intended use.
In essence, FDA’s benefit–Risk assessment is an integrated judgment about the meaningfulness of benefit, the acceptability of risk, and the tolerance of uncertainty.
When seeking approval to market a new drug or to market new uses of an existing drug, a drug company3 must submit a new drug application, containing terabytes of information, including clinical and nonclinical evaluations, manufacturing processes and controls, and proposed product labeling. The application is reviewed by a team representing 10 or more expert disciplines. Each reviewer assesses the adequacy of information, reviews and replicates the data and analyses, and provides recommendations on the approvability of the application from their disciplinary point of view. FDA may also seek outside expertise by convening an Advisory Committee meeting to discuss certain issues in the application. An assigned signatory authority, who is typically the director or deputy director of the clinical office that oversees the relevant therapeutic area, considers the discipline reviews and makes the final decision on FDA’s action—whether to approve the drug application or to issue a “complete response” outlining deficiencies that preclude the application from being approved in its current form. The goal timeframe for FDA’s review and decision-making on a new drug application typically is 6–12 months, depending on the nature of the application.
FDA’s regulatory decisions are based on rigorous scientific assessment, guided by statutory requirements, implemented regulations, and established policy. The criteria for establishing that a drug is “effective” are explicit in statute [FD&C Act Section 505(d)]: the application must demonstrate “substantial evidence that the drug will have the effect it purports or is represented to have under proposed labeled conditions of use,” with FDA outlining the evidentiary standards in its detailed regulations (21CFR 314.125-126). The criteria for establishing that a drug is “safe,” however, are not explicitly defined in statute. Recognizing that almost no drug is safe in the absolute sense (i.e., has no chance of harm), FDA interprets the demonstration of safety as a determination that a drug’s benefits outweigh its risks. This policy interpretation establishes benefit–Risk assessment as a foundation for regulatory approval.
In essence, FDA’s benefit–Risk assessment is an integrated judgment about the meaningfulness of benefit, the acceptability of risk, and the tolerance of uncertainty.4 Benefit–Risk assessment for any drug under review is necessarily case-specific, taking into account specific circumstances and available information. With that said, all benefit–Risk assessments share these overarching elements.
- Therapeutic Context: Sound decisions about the acceptability of a technology are made within a decision context. In the case of drugs, that context is the drug’s proposed use, the patient population’s medical needs, and how well those needs are currently being met. In general, there is a greater tolerance for risk and uncertainty in the context of serious diseases when more effective and well-tolerated therapies are clearly needed. There is generally less tolerance for risk or uncertainty in contexts where several safe and effective treatments are already available and/ or when the drug is intended for mild conditions and generally healthy populations.
- Evidence About the Drug’s Benefits and Risks: FDA’s decisions are informed by evidence from a multitude of sources, including pivotal and supportive clinical trials, toxicology studies, adverse event reporting, patient input, and product quality assessments. Expert reviewers thoroughly evaluate the adequacy of the study design, study conduct, data integrity, and data analysis. They review detailed case reports of serious adverse events along with supporting clinical, pharmacology, and toxicology information to assess potential drug causality.
- Uncertainty: No matter how well clinical evaluations are designed and implemented, some residual uncertainty about a drug’s benefits and risks is inevitable at the time of a new drug approval decision because of the inherent limitations of clinical trials. Examples include the uncertainty about the durability of a drug’s benefit over a longer horizon than was studied in the trial, the potential for rare but serious risks not detected during clinical development, and how well patients and health care providers in the real-world setting will take actions needed to reduce the risk of harm. Many uncertainties can be anticipated and addressed during drug development, for example, by including certain safety assessments. Others become apparent only late in development, such as an unanticipated safety signal (e.g., adverse effects to the heart or liver) that emerges during a trial.
- Regulatory Options to Reduce Risk and Uncertainty: FDA has several tools to facilitate greater assurance of a drug’s favorable bene-fit-risk profile, including requirements for the drug company to conduct additional studies postapproval, requirements on additional warnings and instructions that must be communicated, and requirements for the drug company to ensure that specific risk mitigation measures are put in place within the healthcare system. The extent and nature of any additional requirements are guided by regulatory statute. For example, certain risk mitigation strategies (e.g., ensuring documentation of precautionary blood tests after administering a drug) can only be required by FDA as a condition for approval in cases where FDA deems the risk mitigation necessary to ensure that the drugs benefits outweigh its risks, in other words in “must have” situations rather than “nice to have.”
No matter how well clinical evaluations are designed and implemented, some remaining uncertainty about a drug’s benefits and risks is inevitable at the time of a new drug approval decision because of the inherent limitations of clinical trials.
A benefit–Risk assessment can be straightforward for applications that clearly demonstrate that a drug has meaningful benefits and a safety profile that is well characterized with no serious safety concerns. It becomes more challenging in cases where there are known serious risks, for example, that are life-threatening or damaging to organs, or when there remains significant uncertainty about the potential for serious risks. In such cases, additional assurances might be needed to help ensure a favorable benefit–Risk balance, for example, by narrowing the approved use of the drug for only patients with the most serious forms of disease, or by providing evidence that proposed risk mitigation measures can effectively be implemented in the real-world setting. If there is not sufficient assurance that the drug’s benefits outweigh the risks, then FDA will not approve the drug and will provide input to the company on what further information would be necessary to support future approval.
FDA’s oversight of a new drug does not end with marketing approval. As long as the product is marketed, regulations require the drug company to monitor, evaluate, and report postmarket safety information (21 CFR 314.80). They may also conduct additional studies to evaluate the drug for new uses. In some cases, new evidence might resolve uncertainties about a drug’s safety that had complicated the premarket assessment. Or, new uncertainties can arise with the emergence of surprising risk signals that were not detected in premarket assessment. The therapeutic context also continues to evolve as understanding of disease improves and as new treatments become available. All of these factors inform FDA’s understanding of the drug’s benefit–Risk profile over its lifecycle. Changes in benefit–Risk judgments can lead FDA to determine that new studies, safety labeling information, and/or risk management are necessary, or, rarely, that a favorable benefit–Risk profile is no longer feasible and the drug should be withdrawn from the market.
The ultimate focus of benefit–Risk assessment is the health and well-being of the patients who directly experience the outcomes of treatment. Thus, an adequate understanding of patients’ needs, experiences, and values is critical. This includes, for example, the extent of their unmet medical needs, the clinical benefits that matter most to them, and the benefit and risk tradeoffs they are willing to make. Historically, patient input to inform regulatory decision making was limited. However, in the early 2010s, FDA renewed its commitment to enabling meaningful patient input and, in collaboration with others in public health, academia, and industry, has advanced more inclusive approaches to engage patients and more robust standards and methods to collect scientific data directly from patients that can support regulatory benefit–Risk assessment [8].
FDA’s Benefit–Risk Framework
There are many ways to conduct a pharmaceutical benefit–Risk assessment [9]. Some approaches are qualitative in nature, focused on ensuring a systematic and structured consideration of critical decision factors. Other approaches involve quantitative decision-analytic or modeling techniques to assess benefits, risks, tradeoffs, and uncertainties.
FDA is recognized as a global leader in advancing structured benefit–Risk assessment to meet regulatory needs [10]. FDA implemented its Benefit–Risk Framework in the mid-2010s to promote greater consistency in its approach and clarity of its rationale [11]. Following decision science principles [12], the Benefit–Risk Framework (Figure 1) is a qualitative structure approach to elucidate the most decision-relevant information from a drug review and integrate it into a summary judgment. Its simplicity allows flexibility to capture the nuances of each case-specific assessment.
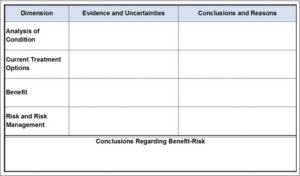
Figure 1.FDA’s Benefit–Risk Framework for new drug review.
FDA’s Benefit–Risk Framework rows outline the high-level decision factors. Analysis of Condition and Current Treatment Options establish the drug’s therapeutic context, serving as the lens by which the nature and importance of the benefits and risks are judged. Benefits and Risks summarize the product-specific information, for example, clinical study findings, that are most pertinent to the benefit–Risk assessment.
The columns distinguish two types of input needed for each factor. Evidence and Uncertainties is a more objective articulation of the facts of the assessment. Conclusions and Reason provides the more subjective judgments of the strength of the evidence and significance of the findings. Explicit splitting of the facts from judgments was an important feature of FDA’s Framework, recognizing the distinct roles of each working in concert.
The final element, Conclusions Regarding Benefit–Risk, is an integrated summary assessment of the most important benefits, risks, and uncertainties, weighed in the context of the severity of the condition and patients’ unmet treatment needs, that supports a conclusion about the drug’s approvability and any special requirements (e.g., risk mitigation). As fundamentally a qualitative tool, the Framework does not require sophisticated numerical techniques to weigh tradeoffs, yet it may include them when feasible and provide specific value (see [13]).
Benefit–Risk Considerations, in More Detail
The simplicity of FDA’s Benefit–Risk Framework in no way diminishes the complexity of the technical and regulatory evaluations that inform it. Within each decision factor exists a multitude of specific considerations. Table 1 is a distillation of factors, uncertainties, and implications generally considered in almost any benefit–Risk assessment. As with any complex technology, the devil is in the details. The purpose of the Framework is to facilitate reviewers in considering and communicating the most important case-specific evidence and conclusions.
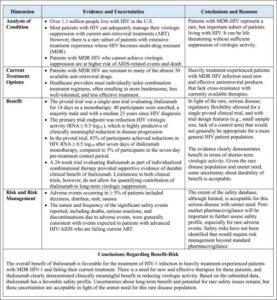
Table 1. General Considerations into FDA’s Benefit–Risk Assessment for New Drug Review [7]
As a less technical illustration, Figure 2 presents a simplified version of a Benefit–Risk Framework [17], [18]5that demonstrates how key factors come together in a summary assessment. In March 2018, FDA approved ibalizumab for the treatment of adults with multidrug-resistant (MDR) HIV-1 infection. This example shows how context matters; although most patients living with HIV can adequately manage their condition with many treatment options, there is a subset of patients who cannot. In the context of this rare, life-threatening form of HIV, FDA tolerated greater uncertainty than it generally would for drugs intended for the wider population of patients with HIV.
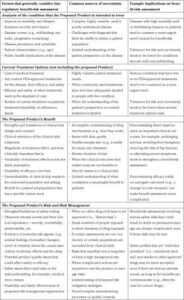
Figure 2.(Simplified) Benefit–Risk Framework: 2018 approval of Ibalizumab for the treatment of multidrug-resistant (MDR) HIV [17], [18].
Societal aspects of benefit–Risk assessment
This commentary has provided an overview of the benefit–Risk assessments that support the regulatory oversight of pharmaceuticals. These assessments follow universal principles of technology governance: clear societal objectives, systematic processes, scientific integrity, uncertainty assessment, risk management, transparency, and adaptive oversight [19]. There are many details and nuances of FDA’s drug regulatory decision making not addressed in this commentary; for a comprehensive treatment, readers are encouraged to explore FDA’s official resources.
Pharmaceutical benefit–Risk assessment inevitably requires resolving, more or less explicitly, several societal questions. One concerns the balance between reducing uncertainty and speeding up patients’ access to treatments for serious and life-threatening diseases. To expedite promising treatments that fill critical unmet needs, FDA implemented an accelerated approval pathway to approve certain drugs based on surrogate measures of benefit, with a requirement that the drug company continue confirmatory trials to establish direct benefit within a certain timeframe after approval [20]. FDA estimates that accelerated approval has enabled access to certain cancer treatments a median of 3 years sooner than traditional approval. However, not all accelerated approvals ultimately produce sufficient confirmatory evidence, as was the case for the drug Makena, which received accelerated approval in 2011 for the prevention of recurrent preterm birth but was ultimately withdrawn from the market in 2023 because it could not confirm meaningful benefit [21].
At times, FDA’s benefit–Risk assessments require judgment of whether patients can sufficiently understand complex risks or take the steps needed to manage them.
Another inherent challenge is the fact that some patients may disproportionally experience benefits, and some patients will disproportionally experience harms, for reasons that may never be completely known. Even favorable benefit–Risk assessments must recognize that in some cases, there will inevitably be therapeutic winners and losers. This requires a dual need by FDA to reduce (through postmarket requirements) resolvable uncertainties and to ensure (through product labeling) that patients and their healthcare providers have the information they need to make the most informed decisions.
There are also times when pharmaceutical benefits and risks extend beyond the patient. Consider, for example, a pregnant woman who requires a treatment that can pose risks to their fetus; the societal protection against infectious diseases offered by vaccines; and the fact that opioid medications required by many patients to manage severe pain confer a risk of overdose to others who may access them. While the essence of a benefit–Risk assessment is the patients for whom the drug is intended, FDA acknowledges that, when relevant, these broader public health considerations become important factors [7, p.5].
At times, FDA’s benefit–Risk assessments require judgment about whether patients can sufficiently understand complex risks or take the steps needed to manage them. This has revealed itself in some of FDA’s decisions about prescription drugs, for example, by requiring that a female patient take a pregnancy test in a medical setting every month before receiving an acne medication that carries the risk of fetal toxicities. And it is always a consideration when determining whether a prescription drug can switch to over-the-counter status. Even when it initially takes a more precautionary stance, FDA has demonstrated a willingness to reassess as more behavioral insight is gained (see [22]) or as new decision support tools are available [23].
Ultimately, these challenges point to a perhaps more fundamental question: whose perspective should weigh more in making difficult judgments about establishing benefits, accepting risk, and tolerating uncertainty? Is it that of the regulator who has made the expert scientific assessment and who bears responsibility to protect public health, or that of the patient who bears directly the outcomes of disease and treatment? FDA acknowledges that “at times, there may be a tension” between its need to consider health at a population-level and the assessments made by patients and providers that consider their individual-level circumstances and values [7, p. 5]. With that said, within its role as a gatekeeper to the public’s pharmaceutical access, FDA has demonstrated a commitment, through programs such as Expanded Access [24], to allow patients whohave significant unmet needs the opportunity to individually access certain drugs that have not been approved.
These societal complexities further exemplify the need for sound regulatory assessments that are grounded in thorough science, societal (patient) input, principled judgment, transparent rationale, and adaptive oversight. FDA’s Benefit–Risk Framework can support all of these needs.
ACKNOWLEDGMENTS
I am a former employee of the U.S. Food and Drug Administration. The views expressed in this article are those of the author and should not be interpreted as representing FDA policies or positions.
Author Information
Sara L. Eggers is a decision scientist specializing in regulatory decision making. She is the former director of the Decision Support and Analysis Staff at the U.S. Food and Drug Administration’s Center for Drug Evaluation and Research. Dr. Eggers has a PhD in engineering and public policy from Carnegie Mellon University, Pittsburgh, PA, USA. Email: saralynneggers@gmail.com.
____________________
To read the full version of this article, including references, click HERE.
___________________
 JOIN SSIT
JOIN SSIT